Characterization of proteins and post-translational modifications by liquid-chromatography mass spectrometry
Over the past three decades, mass spectrometry and the combination of liquid-chromatography with mass spectrometry (LC-MS) became one of the most powerful techniques for the identification and characterization of proteins and post-translational modifications. While originally triple quadrupole mass spectrometers have been used, these have later usually been replaced first by ion traps and later by either QTOF or Orbitrap technology based systems because of their higher resolution, speed and mass accuracy.
Mass spectrometry and proteomics became a dominant area of research which deals with high-throughput protein identification, analysis of isoforms and modifications, alternative gene products, disease origins, etc.
Also, many aspects of critical quality attributes (CQA) of biologics are nowadays investigated by LC-MS and protein mass spectrometry.
How can mass spectrometry help in the analysis of proteins?
Proteins consist out of a usually linear sequence of 22 amino acids (actually more if some specific variants are counted as well). Most of these amino acids have distinct elemental compositions and molecular masses.
When a protein is synthesized, these amino acids assemble into a chain where the mass is determined by the masses of the amino acids from which it is formed. Comparing the measured mass of the intact protein can tell us whether there is a difference between the mass we calculated from the amino acid composition or not. In the first case either the expected compostion is wrong or a so-called post-translational modification introduced a change after the synthesis of the protein chain.
The analysis of the intact protein by mass spectrometry can provide us with a first information about the integrity of the protein.
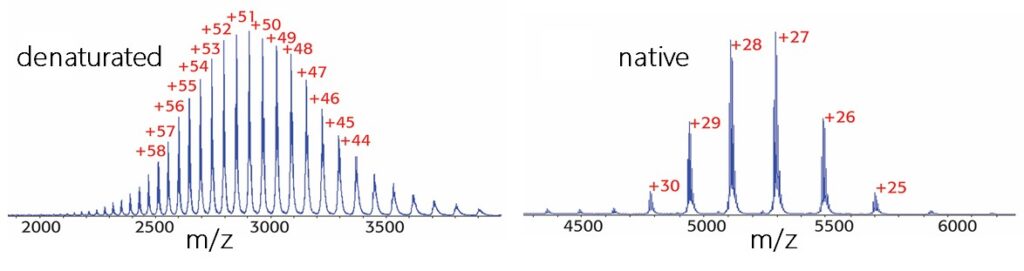
Some post-translational modifications can also be heterogeneous, such as glycosylations. These modifcations then also introduce a heterogeneity into the intact modified protein so that multiple masses for one protein may appear in the mass spectrum. A typical example for this are monoclonal antibodies which carry (at least) a conserved glycosylation at both heavy chains.
Mass spectrometric analysis of a monoclonal antibody under denaturated and native mass spectrometry conditions. The heterogeneity in the individual charge states is caused by the glycosylation on the heavy chain.
While for biologics the protein sequence is usually known, for proteins from biological samples it might not be known. This is the area of classical proteomics analysis using protein identification by by mass spectrometry.
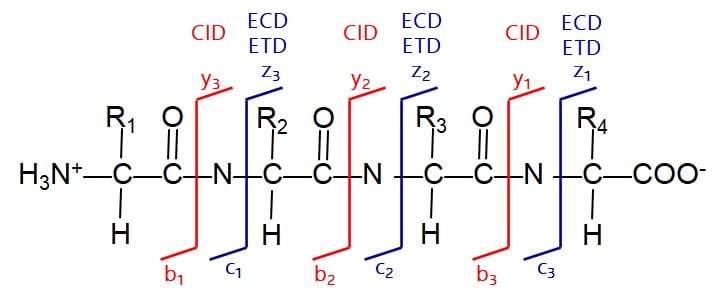
Here the intact protein is commonly digested using specific enzymes (most commonly Trypsin), which cleave the protein chain at specific residues int smaller, more handy peptides. These peptides are then separated by liquid chromatography and fragmented in the mass spectrometer. Under collision-induced dissociation conditions, the fragmentation of the peptide chain usually always occurs within the amide bond between the amino acid residues. As a result we obtain a spectrum with specific peaks showing a spacing which corresponds to the amino acids forming the peptide on our triple quadrupole-, QTOF- or Orbitrap-based mass spectrometer. With a set of peptides and the amino acid sequence derived from their MS/MS spectra we can now run a database search to identify the protein based on a sequence originally obtained e.g. from genetic sequencing and being stored in databases.
Fragmentation of a peptide chain along the sequence with fragment ions origination either from collision-induced dissociation (CID) or electron-capture (ECD) or electron-transfer dissociation (ETD)
How can MS Vision help you with that?
MS Vision is specialized on liquid chromatography-mass spectrometry (LC-MS) technology. We offer dedicated services around this technology like
We gebruiken cookies om onze site goed te laten functioneren. Wanneer je op ACCEPT klik ga je er mee akkoord dat we de cookies gebruiken.
For proper functionality of our website we use Cookies. By clicking on ACCEPT you agree with the usage of these Cookies.Cookie settingsACCEPT
Manage consent
Privacy Overview
This website uses cookies to improve your experience while you navigate through the website. Out of these, the cookies that are categorized as necessary are stored on your browser as they are essential for the working of basic functionalities of the website. We also use third-party cookies that help us analyze and understand how you use this website. These cookies will be stored in your browser only with your consent. You also have the option to opt-out of these cookies. But opting out of some of these cookies may affect your browsing experience.
Necessary cookies are absolutely essential for the website to function properly. This category only includes cookies that ensures basic functionalities and security features of the website. These cookies do not store any personal information.
Any cookies that may not be particularly necessary for the website to function and is used specifically to collect user personal data via analytics, ads, other embedded contents are termed as non-necessary cookies. It is mandatory to procure user consent prior to running these cookies on your website.